
From a young age, Galila Yohannes began to show signs of a visual field disorder that impaired her ability to function independently until further deterioration began and she almost lost her sight.
Now aged six, she was taken by her worried parents – foreign workers who came from Eritrea – to the outpatient eye institute at Tel Aviv Sourasky Medical Center and after a long series of tests at a pediatric retina clinic, Galila was diagnosed with a rare hereditary degenerative disease called retinitis pigmentosa (RP).
Dr. Shulamit Schwartz, a senior ophthalmologist and director of the Unit for Advanced Technologies in Retinal Surgery, and retina specialist Prof. Dina Zur, explained: “Retina degeneration is caused by genetic mutations that prevent the production of a protein required for vision. In Galila’s case, it is a disease known as retinitis pigmentosa that already at a young age causes a disturbance in the field of vision, night vision and later also in visual acuity up to blindness. Galila was diagnosed with the same mutation in the same rare gene.”
What is retinis pigmentosa?
The retina is a thin piece of tissue lining the back of the eye. Rod and cone photoreceptors in the retina convert light into electrical signals that the brain interprets as vision. People with RP suffer from a gradual decline in their vision, because photoreceptors degenerate. Forms of RP and related diseases include Usher syndrome, Leber congenital amaurosis, and Bardet-Biedl syndrome, among others.
RP, typically diagnosed in children, adolescents and young adults, is a progressive disorder. The rate of progression and degree of visual loss varies from person to person. Many people with RP are legally blind by age 40, with a central visual field of less than 20 degrees in diameter. Genetic mutations can be passed from parent to offspring through one of three genetic inheritance patterns. One in every 4,000 Americans and about one in 5,000 people worldwide have RP, making it the most common inherited disease of the retina.
RP is typically diagnosed in children, adolescents and young adults. It is a progressive disorder. The rate of progression and degree of visual loss varies from person to person. Many people with RP are legally blind by age 40, with a central visual field of less than 20 degrees in diameter Genetic mutations can be passed from parent to offspring through one of three genetic inheritance patterns. One in every 4,000 Americans and about one in 5,000 worldwide have RP, making it the most common inherited disease of the retina.
In recent years, the first gene therapy for this extremely rare disease caused by a mutation in the "65RPE" gene was registered with the Health Ministry for this condition, and it was added to the drug basket after it had already been approved earlier in the US Food and Drug Administration (2017) and Europe.
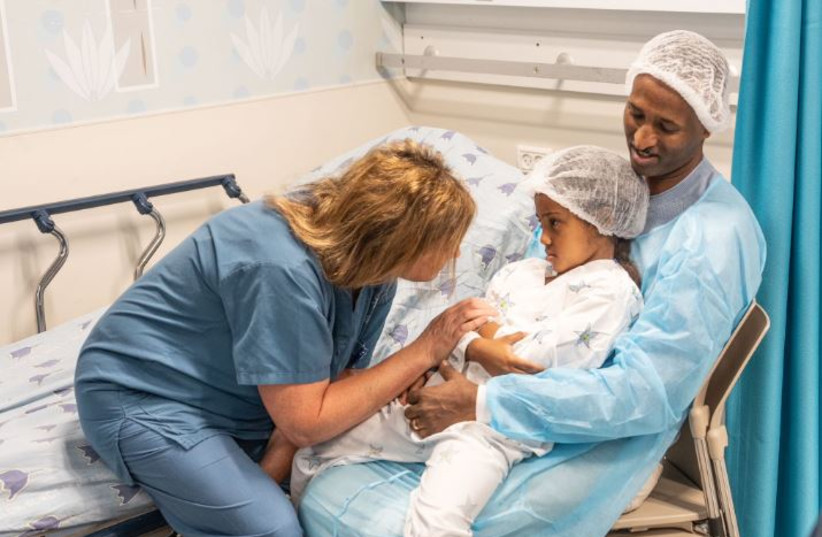
“The doctors explained to us that Galila would have to undergo surgery with an injection of the drug under the retina in both eyes so she would be able to see again,” her parents recalled. “We were very worried, but we couldn’t bear the situation affecting our sweet Galila. We put all our trust in the amazing doctors in the hospital. We had the feeling that they would do everything to restore her sight and help her return to seeing and living like any other child her age.”
Under the experienced hands of the skilled team of ophthalmologists, led by Schwartz, Zur and Prof. Adiel Barak, the unique procedure was performed for the first time at Sourasky. She was injected with the innovative drug called Luxturna (voretigene neparvovec-rzyl), a prescription gene therapy product used for the treatment of patients with inherited retinal disease due to mutations in both copies of the RPE65 gene, which can be confirmed only by genetic testing. The drug is injected in each eye separately and in a one-time fashion directly under the retina and makes possible the transfer of a normal copy of the gene to the retinal cells so they can produce the missing protein needed for vision.
This week, Galila came with her parents for a check-up after the surgeries on both eyes and the tests showed an improvement in visual function. “Her condition is expected to improve over time, and we anticipate that Galila will be able to perform daily tasks more independently, which she could not do before the treatment.”
Prof. Hagit Baris Feldman, director of the hospital’s genetics institute, and Dr. Dafna Marom, deputy director of the institute and coordinator of the field of pediatric genetics, explained: “Genetic tests are available for patients with hereditary retinal degeneration; they are vital for identifying the defective gene that causes the disease. Finding the gene can lead to treatment of the disease, as in the case of Galila or allow participation in future clinical studies